
Nonadiabatic dynamics of floppy hydrogen bonded complexes: the case of the ionized ammonia dimer†
Abstract
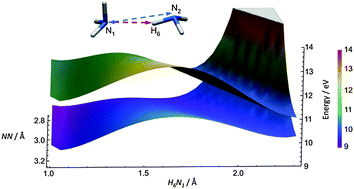
In the case of the ammonia dimer, we address the following questions: how ultrafast ionization dynamics is controlled by hydrogen bonding and whether we can control the products via selective ionization of a specific electron. We use quantum chemical calculations and ab initio non-adiabatic molecular dynamics simulations to model the femtosecond dynamics of the ammonia dimer upon ionization. The role of nuclear quantum effects and thermal fluctuations in predicting the structure of the dimer is emphasized; it is shown that the minimum energy and vibrationally averaged structures are rather different. The ground state structure subsequently controls the ionization dynamics. We describe reaction pathways, electronic population transfers and reaction yields with respect to ionization from different molecular orbitals. The simulations showed that the ionized ammonia dimer is highly unstable and its decay rate is primarily driven by the position of the electron hole. In the case of ground state ionization (i.e. the HOMO electron is ionized), the decay is likely to be preceded by a proton transfer (PT) channel yielding NH4+ and NH2˙ fragments. The PT is less intense and slower compared with the ionized water dimer. After ionizing deeper lying electrons, mainly NH3+˙ and NH3 fragments are formed. Overall, our results show that the ionization dynamics of the ammonia and water dimers differ due to the nature of the hydrogen bond in these systems.
 Please wait while we load your content...
Please wait while we load your content...

