
Pressure-induced structural and valence transition in AgO
Abstract
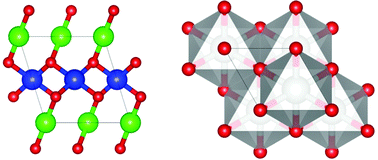
The pressure-induced evolution of AgO crystal structures and the oxygen environment of Ag atoms were investigated by means of density functional theory with a hybrid functional and a structure prediction method. Under ambient conditions, AgO has two nonequivalent Ag1 and Ag2 sites that adopt linear and square planar oxygen environment configuration, respectively, corresponding to Ag mixed-valence states. The results show that both the coordination environment and the valence state of the Ag1 site are sensitive to pressure and will gradually approach those of the Ag2 site as it increases. The band gap also decreases significantly and at 75 GPa AgO experiences a pressure-induced semiconductor-to-metal transition. At ∼77 GPa, there is a structural transition from monoclinic (P21/c) to trigonal (R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m), accompanied by a valence state transition from the mixed-valence state to a single-valence state.
m), accompanied by a valence state transition from the mixed-valence state to a single-valence state.
 Please wait while we load your content...
Please wait while we load your content...

