
Toward polarizable AMOEBA thermodynamics at fixed charge efficiency using a dual force field approach: application to organic crystals
Abstract
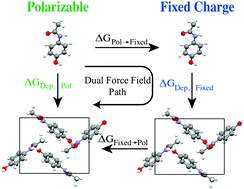
First principles prediction of the structure, thermodynamics and solubility of organic molecular crystals, which play a central role in chemical, material, pharmaceutical and engineering sciences, challenges both potential energy functions and sampling methodologies. Here we calculate absolute crystal deposition thermodynamics using a novel dual force field approach whose goal is to maintain the accuracy of advanced multipole force fields (e.g. the polarizable AMOEBA model) while performing more than 95% of the sampling in an inexpensive fixed charge (FC) force field (e.g. OPLS-AA). Absolute crystal sublimation/deposition phase transition free energies were determined using an alchemical path that grows the crystalline state from a vapor reference state based on sampling with the OPLS-AA force field, followed by dual force field thermodynamic corrections to change between FC and AMOEBA resolutions at both end states (we denote the three step path as AMOEBA/FC). Importantly, whereas the phase transition requires on the order of 200 ns of sampling per compound, only 5 ns of sampling was needed for the dual force field thermodynamic corrections to reach a mean statistical uncertainty of 0.05 kcal mol−1. For five organic compounds, the mean unsigned error between direct use of AMOEBA and the AMOEBA/FC dual force field path was only 0.2 kcal mol−1 and not statistically significant. Compared to experimental deposition thermodynamics, the mean unsigned error for AMOEBA/FC (1.4 kcal mol−1) was more than a factor of two smaller than uncorrected OPLS-AA (3.2 kcal mol−1). Overall, the dual force field thermodynamic corrections reduced condensed phase sampling in the expensive force field by a factor of 40, and may prove useful for protein stability or binding thermodynamics in the future.
- This article is part of the themed collection: Insights from advanced methods in molecular dynamics
 Please wait while we load your content...
Please wait while we load your content...

