
Understanding the anomalous behavior of Vegard's law in Ce1−xMxO2 (M = Sn and Ti; 0 < x ≤ 0.5) solid solutions†
Abstract
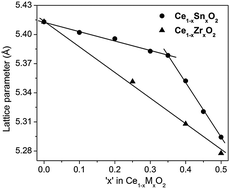
The dependence of the lattice parameter on dopant concentration in Ce1−xMxO2 (M = Sn and Ti) solid solutions is not linear. A change towards a steeper slope is observed around x ∼ 0.35, though the fluorite structure (space group Fm3m) is preserved up to x = 0.5. This phenomenon has not been observed for Ce1−xZrxO2 solid solutions showing a perfectly linear decrease of the lattice parameter up to x = 0.5. In order to understand this behavior, the oxidation state of the metal ions, the disorder in the oxygen substructure and the nature of metal–oxygen bonds have been analyzed by XPS, 119Sn Mössbauer spectroscopy and X-ray absorption spectroscopy. It is observed that the first Sn–O coordination shell in Ce1−xSnxO2 is more compact and less flexible than that of Ce–O. The Sn coordination remains symmetric with eight equivalent, shorter Sn–O bonds, while Ce–O coordination gradually splits into a range of eight non-equivalent bonds compensating for the difference in the ionic radii of Ce4+ and Sn4+. Thus, a long-range effect of Sn doping is hardly extended throughout the lattice in Ce1−xSnxO2. In contrast, for Ce1−xZrxO2 solid solutions, both Ce and Zr have similar local coordination creating similar rearrangement of the oxygen substructure and showing a linear lattice parameter decrease up to 50% Zr substitution. We suggest that the localized effect of Sn substitution due to its higher electronegativity may be responsible for the deviation from Vegard's law in Ce1−xSnxO2 solid solutions.
 Please wait while we load your content...
Please wait while we load your content...

