
Polymorphism in α-sexithiophene crystals: relative stability and transition path†
Abstract
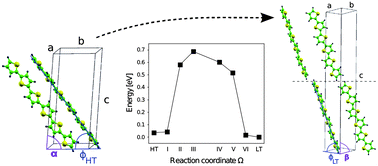
We present a joint theoretical and experimental study to investigate polymorphism in α-sexithiophene (6T) crystals. By means of density-functional theory calculations, we clarify that the low-temperature phase is favorable over the high-temperature one, with higher relative stability up to 50 meV per molecule. This result is in agreement with our thermal desorption measurements. We also propose a transition path between the high- and low-temperature 6T polymorphs, estimating an upper bound for the energy barrier of about 1 eV per molecule. The analysis of the electronic properties of the investigated 6T crystal structures complements our study.
 Please wait while we load your content...
Please wait while we load your content...

