
How molecular is the chemisorptive bond?†
Abstract
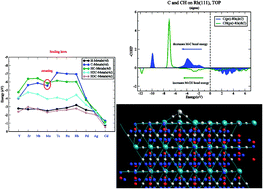
Trends in adsorption energies as a function of transition metal differ for adsorbates that are attached atop a surface atom or are adsorbed onto a high coordination site. When adsorption onto early and late transition metals is compared variation in relative bond energies of adsorbates attached to different sites is large. A theoretical understanding is provided based on the analysis of the electronic structure of the respective chemical bonds. The electronic structure analysis is based on partial density of states (PDOS) and bond order overlap population densities from crystal orbital Hamiltonian population (COHP) calculations available from DFT electronic structure computations. This is complemented by calculations of Bader charge densities and electron density topology properties. Variation of the respective bond energies depends on the symmetry of the molecular orbitals that form the chemical bond. The key electronic structure parameters are the position of the Fermi level in the bonding or antibonding molecular orbital partial density of states region of the chemical bond and chemical bond polarity. These are very different for adsorbates adsorbed onto the same transition metal surface, but which have different coordination with surface metal atoms. The adsorption energies and the respective electronic structures of adatoms H, C and O and molecular fragments CHx (x = 1–3) are compared with those of the analogous molecules that contain a single transition metal atom. When adsorbed atop, trends in bond energies are remarkably similar to those of the corresponding molecules. The difference in bond energies of adsorbates and transition metal molecules, i.e. the embedding energy, is shown to consist of three contributions: quenching of the sometimes high molecular spin states, weakening of the adsorbate–surface interaction energy and weakening of the metal–metal atom bond energies next to the adsorbate. Conventional scaling rules of the interaction energies of adsorbed CHx (0 < x ≤ 3) fragments are satisfied only for adsorbates in high coordination sites. For the early transition metals a breaking of this rule is found for C and CH or N and NH when adsorbed atop a transition metal surface or when they are part of a transition metal molecule. The M–C bond energy is found to be only stronger than that of the M–CH bond as long as the Fermi level or the HOMO is located in the antibonding molecular orbital partial density of states of the chemical bond.
- This article is part of the themed collections: PCCP Perspectives and Developments in Density Functional Theory
 Please wait while we load your content...
Please wait while we load your content...

