
How protonation and deprotonation of 9-methylguanine alter its singlet O2 addition path: about the initial stage of guanine nucleoside oxidation†
Abstract
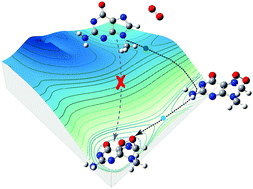
Mutagenicity of singlet O2 is due to its oxidatively generated damage to the guanine nucleobases of DNA. Oxidation of neutral guanosine has been assumed to be initiated by the formation of a transient 4,8-endoperoxide via a Diels–Alder cycloaddition of singlet O2. Protonation and deprotonation of guanosine represent another factor related to DNA damage and repair. Herein, 9-methylguanine was utilized as a model substrate to mimic the correlation between singlet O2 oxidation of the nucleoside and its ionization states, both in the absence and in the presence of water ligands. We used guided-ion-beam scattering tandem mass spectrometry to detect and quantify transient intermediates at room temperature. To provide a reliable description of reaction potential surfaces, different levels of theory including restricted and unrestricted density functional theory, CCSD(T), MP2, and multi-reference CASSCF and CASMP2 were applied. By means of molecular potential, kinetic and direct dynamics simulations, two reaction pathways were identified and neither follows the mechanism for neutral guanosine. Singlet O2 oxidation of protonated 9-methylguanine begins by a concerted cycloaddition; but it is mediated by a 5,8-endoperoxide. By contrast, a concerted cycloaddition does not occur for deprotonated 9-methylguanine. The latter involves a stepwise addition starting with the formation of an 8-peroxide, which subsequently evolves to a 4,8-endoperoxide. This dichotomy implies that acidic and basic media may lead to different chemistries for guanosine oxidation in aqueous solutions, starting from initial stage. The comparison with oxidation of protonated/deprotonated guanine illustrates the different mechanisms and products and particularly the suppressed oxidizability of 9-methylguanine vs. free guanine.
 Please wait while we load your content...
Please wait while we load your content...

