
Abstract
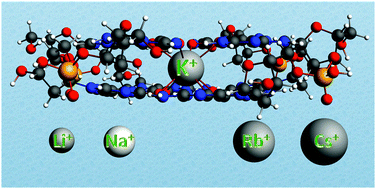
The alkali metal ion affinity of guanine quadruplexes has been studied using dispersion-corrected density functional theory (DFT-D). We have done computational investigations in aqueous solution that mimics artificial supramolecular conditions where guanine bases assemble into stacked quartets as well as biological environments in which telomeric quadruplexes are formed. In both cases, an alkali metal cation is needed to assist self-assembly. Our quantum chemical computations on these supramolecular systems are able to reproduce the experimental order of affinity of the guanine quadruplexes for the cations Li+, Na+, K+, Rb+, and Cs+. The strongest binding is computed between the potassium cation and the quadruplex as it occurs in nature. The desolvation and the size of alkali metal cations are thought to be responsible for the order of affinity. Until now, the relative importance of these two factors has remained unclear and debated. By assessing the quantum chemical ‘size’ of the cation, determining the amount of deformation of the quadruplex needed to accommodate the cation and through the energy decomposition analysis (EDA) of the interaction energy between the cation and the guanines, we reveal that the desolvation and size of the alkali metal cation are both almost equally responsible for the order of affinity.
- This article is part of the themed collection: Developments in Density Functional Theory
 Please wait while we load your content...
Please wait while we load your content...

