
Control of valence and conduction band energies in layered transition metal phosphates via surface functionalization†
Abstract
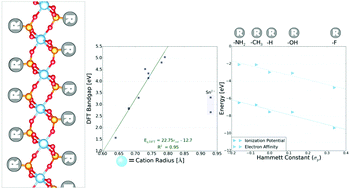
Layered transition metal phosphates and phosphites (TMPs) are a class of materials composed of layers of 2D sheets bound together via van der Waals interactions and/or hydrogen bonds. Explored primarily for use in proton transfer, their unique chemical tunability also makes TMPs of interest for forming large-scale hybrid materials. Further, unlike many layered materials, TMPs can readily be solution exfoliated to form single 2D sheets or bilayers, making them exciting candidates for a variety of applications. However, the electronic properties of TMPs have largely been unstudied to date. In this work, we use first-principles computations to investigate the atomic and electronic structure of TMPs with a variety of stoichiometries. We demonstrate that there exists a strong linear relationship between the band gap and the ionic radius of the transition metal cation in these materials, and show that this relationship, which opens opportunities for engineering new compositions with a wide range of band gaps, arises from constraints imposed by the phosphorus–oxygen bond geometry. In addition, we find that the energies of the valence and conduction band edges can be systematically tuned over a range of ∼3 eV via modification of the functional group extending from the phosphorus. Based on the Hammett constant of this functional group, we identify a simple, predictive relationship for the ionization potential and electron affinity of layered TMPs. Our results thus provide guidelines for systematic design of TMP-derived functional materials, which may enable new approaches for optimizing charge transfer in electronics, photovoltaics, electrocatalysts, and other applications.
 Please wait while we load your content...
Please wait while we load your content...

