
A spectroscopic and theoretical study in the near-infrared region of low concentration aliphatic alcohols†
Abstract
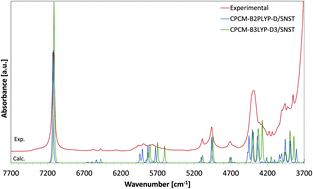
The near-infrared (NIR) spectra of low-concentration (5 × 10−3 M) solutions in CCl4 of basic aliphatic alcohols, methanol, ethanol, and 1-propanol were, for the first time, calculated by second-order vibrational perturbation theory computations and were compared with the corresponding experimental data. Density functional theory (DFT) using single hybrid (B3LYP) and double hybrid (B2PLYP) density functionals and their derivatives with additional empirical dispersion correction (B3LYP-D3 and B2PLYP-D, respectively) and second order Møller–Plesset perturbation theory were used in combination with selected basis sets including fairly new basis sets from the “spectroscopic” SNS family, double-ζ SNSD and triple-ζ SNST basis sets. Each time, anharmonic vibrational modes and intensities were calculated by using second-order vibrational perturbation theory. The effect of solvent cavity on the calculated results was included by the application of a self-consistent reaction field with a polarized continuum model. Ethanol and 1-propanol have conformational isomerism; following a conformational analysis, theoretical spectra of all isomers were calculated and their final predicted NIR spectra were obtained as Boltzmann-averaged spectra of resolved conformers. For ethanol and 1-propanol, the observed broadening of the overtone band of the OH stretching mode was well reflected by the differences in the position of the relevant band among conformational isomers of these alcohols; the effect of solvent on broadening was also discussed. Detailed band assignments in the experimental NIR spectra of the studied alcohols were proposed based on the calculation of potential energy distributions. The final accuracy of the predicted NIR spectra for each of the theoretical methods was estimated based on the errors in calculated frequencies of overtones and combination bands.
 Please wait while we load your content...
Please wait while we load your content...

