
Kohn–Sham calculations of NMR shifts for paramagnetic 3d metal complexes: protocols, delocalization error, and the curious amide proton shifts of a high-spin iron(ii) macrocycle complex†
 a
and
a
and
Abstract
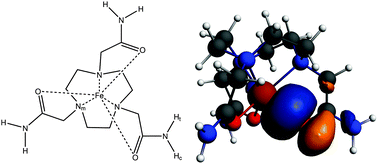
A theory for the nuclear chemical shifts of molecules in arbitrary spin states is applied to a set of paramagnetic organometallic complexes of 3d metals. Ligand chemical shifts are calculated and analyzed using Kohn–Sham (KS) density functional theory with and without relativistic corrections. The roles of the KS delocalization error, Gaussian-type versus Slater-type basis sets, relativistic effects (scalar and spin–orbit), and zero field splitting (ZFS) are investigated. A strong functional dependence of the chemical shifts is apparent and correlated with the delocalization error. The functional dependence is between one and two orders of magnitude larger than variations of the NMR shifts due the other influences that are investigated. ZFS effects are negligible in the determination of the NMR chemical shifts of the complexes except at very low temperatures. The DFT calculated shifts agree reasonably well with experiment. A 73 ppm difference in the NMR shifts of the two protons in the amide groups of a high-spin Fe(II) macrocycle complex arises from selective O → Fe dative bonding that only involves the transfer of β spin density, along with orbital delocalization throughout the ligand bonding framework which electronically couples the coordinating oxygen lone pair orbitals directly to the amide trans proton.
- This article is part of the themed collection: Developments in Density Functional Theory
 Please wait while we load your content...
Please wait while we load your content...

