
A multi-level quantum mechanics and molecular mechanics study of SN2 reaction at nitrogen: NH2Cl + OH− in aqueous solution
Abstract
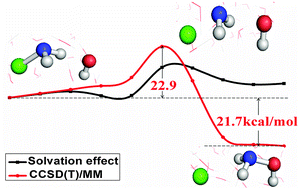
We employed a multi-level quantum mechanics and molecular mechanics approach to study the reaction NH2Cl + OH− in aqueous solution. The multi-level quantum method (including the DFT method with both the B3LYP and M06-2X exchange–correlation functionals and the CCSD(T) method, and both methods with the aug-cc-pVDZ basis set) was used to treat the quantum reaction region in different stages of the calculation in order to obtain an accurate potential of mean force. The obtained free energy activation barriers at the DFT/MM level of theory yielded a big difference of 21.8 kcal mol−1 with the B3LYP functional and 27.4 kcal mol−1 with the M06-2X functional respectively. Nonetheless, the barrier heights become very close when shifted from DFT to CCSD(T): 22.4 kcal mol−1 and 22.9 kcal mol−1 at CCSD(T)(B3LYP)/MM and CCSD(T)(M06-2X)/MM levels of theory, respectively. The free reaction energy obtained using CCSD(T)(M06-2X)/MM shows an excellent agreement with the one calculated using the available gas-phase data. Aqueous solution plays a significant role in shaping the reaction profile. In total, the water solution contributes 13.3 kcal mol−1 and 14.6 kcal mol−1 to the free energy barrier heights at CCSD(T)(B3LYP)/MM and CCSD(T)(M06-2X)/MM respectively. The title reaction at nitrogen is a faster reaction than the corresponding reaction at carbon, CH3Cl + OH−.
 Please wait while we load your content...
Please wait while we load your content...

