
Observation and modeling of conformational molecular structures driving the self-assembly of tri-adamantyl benzene on Ag(111)†

Abstract
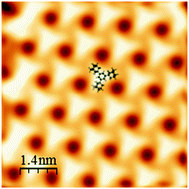
The self-organization of tri-adamantyl (TAB) benzene molecules has been investigated using low temperature scanning tunneling microscopy (LT-STM). The molecular structures have also been studied using molecular modeling. In particular, these calculations have been performed on large areas (1000 nm2) from the atomic structure of the molecular building block, combining molecular dynamics (MD) and Monte-Carlo (MC) approaches. These investigations show that the structure of the molecule and its flexibility allow for the formation of different networks as a function of surface coverage. The calculations demonstrate that the stability of the largest structures is obtained through the increase of the interfacial energy induced by the rotation of the adamantyl groups, a behavior whose consequences explain the subtle contrasts observed in the experimental STM images.
 Please wait while we load your content...
Please wait while we load your content...

