
PPARγ helix 12 exhibits an antagonist conformation†

Abstract
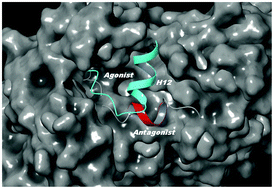
Although the peroxisome proliferator-activated receptor γ (PPARγ) is one of the most studied nuclear receptors (NR), it is still unknown whether its activation helix (helix 12, H12) could exhibit an antagonist conformation as previously demonstrated for most of the NRs. The high H12 flexibility in the apo PPARγ form and the lack of appropriate antagonist ligands complicate the structural and dynamics description by most of the experimental techniques. Based on intensive (≈12 μs) accelerated molecular dynamics (aMD) simulations together with metadynamics and conventional MD runs, we reveal that H12 could exist in an antagonist conformation. This H12 state and the well-known agonist configuration have virtually identical free energy. Notably, significant deviations in the H12 conformations are detected in a homodimer. In chain A the activation helix is stabilized only in a full agonist conformation whereas in chain B, due to agonist to antagonist state exchanges, H12 is oriented toward helix 4. In summary, the results provide an explanation of the observed asymmetry in most of the PPARγ homodimer crystal structures. They also suggest selection guidance for protein moieties and structure candidates that would best serve as potential ligand binding sites to achieve a stable antagonist form of the receptor.
 Please wait while we load your content...
Please wait while we load your content...

