
Unravelling the hydrogen absorption process in Pd overlayers on a Au(111) surface†
Abstract
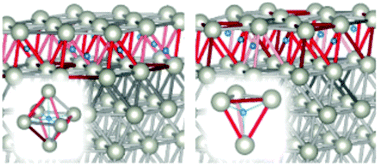
The hydrogen absorption into overlayers of Pd deposited on Au(111) has been investigated by density functional theory (DFT). Hydrogen concentrations, absorption environments, and geometrical and electronic effects have been analyzed, seeking for a better understanding of the general principles governing the process and the effect of foreign supports. The results show that the absorption is more favored than in pure Pd leading to lower absorption energies and less repulsive interactions due to the surface expansion induced by the gold larger lattice constant. Our findings also suggest that the hydrogen absorption process is more favorable for a less number of Pd overlayers. This situation changes gradually until the substrate influence is no longer detected and the pure palladium nature appears. An entangled combination of repulsive forces, strain effect, structural ordering and chemical affinity has been found. The kinetics of hydrogen absorption has been studied as well. Two cases were explored: (1) the absorption of an adsorbed hydrogen atom and (2) the bond-breaking and penetration of a H2 molecule.
 Please wait while we load your content...
Please wait while we load your content...

