
Flatbands in 2D boroxine-linked covalent organic frameworks†
Abstract
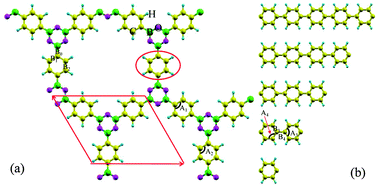
Density functional calculations have been performed to analyze the electronic and mechanical properties of a number of 2D boroxine-linked covalent organic frameworks (COFs), which are experimentally fabricated from di-borate aromatic molecules. Furthermore, the band structures are surprising and show flat-band characteristics which are mainly attributed to the delocalized π-conjugated electrons around the phenyl rings and can be better understood within aromaticity theories. Next, the effects of branch sizes and hydrostatic strains on their band structures are systematically considered within generalized gradient approximations. It is found that their band gaps will start to saturate when the branch size reaches 9. For boroxine-linked COFs with only one benzene ring in the branch, the band gap is robust under compressive strain while it decreases with the tensile strain increasing. When the branch size is equal or greater than 2, their band gaps will monotonously increase with the strain increasing in the range of [−1.0, 2.0] Å. All boroxine-linked COFs are semiconductors with controllable band gaps, depending on the branch length and the applied strain. In comparison with other 2D materials, such as graphene, hexagonal boron nitride, and even γ-graphyne, all boroxine-linked COFs are much softer and even more stable. That is, they can maintain the planar features under a larger compressive strain, which means that they are good candidates in flexible electronics.
 Please wait while we load your content...
Please wait while we load your content...

