
Spotting and designing promiscuous ligands for drug discovery†
Abstract
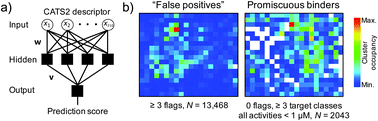
The promiscuous binding behavior of bioactive compounds forms a mechanistic basis for understanding polypharmacological drug action. We present the development and prospective application of a computational tool for identifying potential promiscuous drug-like ligands. In combination with computational target prediction methods, the approach provides a working concept for rationally designing such molecular structures. We could confirm the multi-target binding of a de novo generated compound in a proof-of-concept study relying on the new method.
 Please wait while we load your content...
Please wait while we load your content...

