
An optimization of the LC-MS/MS workflow for deep proteome profiling on an Orbitrap Fusion†
Abstract
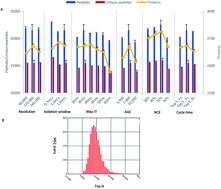
The development of high-resolution mass spectrometers (MS) has greatly advanced the system-wide proteomic profiling and protein post-translational modification (PTM) studies. However, in contrast to current genomic sequencing technologies, huge time cost and laborious workload are the major bottlenecks of current MS-based proteomic approaches for large-scale in-depth proteome sequencing of biological samples. Here we present a stepwise optimization of MS parameters and an off-line reverse phase HPLC fractionation method in the first tribrid MS platform—Orbitrap Fusion, which integrates quadrupole, ultrahigh field Orbitrap and linear ion trap mass analyzers. With off-line high pH separation, we identified more than 5000 proteins using a regular short reverse phase C18 column (10 cm × 75 μm, 3 μm particle size) in a single one hour LC-MS run and 8493 proteins with 6 orders of magnitude of dynamic range in only 10 hour MS running time. Our study provided a fast, cost-efficient and amenable method for deep proteomic analysis and quantification of large-scale biological samples. Significantly, this strategy would facilitate the proteomic disease biomarker discovery.
 Please wait while we load your content...
Please wait while we load your content...

