
Computational studies on the structural, electronic and optical properties of graphene-like MXenes (M2CT2, M = Ti, Zr, Hf; T = O, F, OH) and their potential applications as visible-light driven photocatalysts†
Abstract
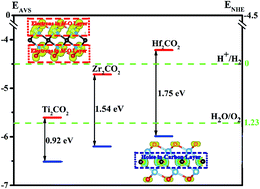
By using the density functional theory (DFT) method, we theoretically investigated the structural, electronic and optical properties of functionalized MXenes with three different geometries, namely M2CT2-I, M2CT2-II and M2CT2-III (M = Ti, Zr, Hf; T = F, O, OH). Except for the M2CO2-II and M2CO2-III, there is no negative frequency in the phonon spectra of M2CT2 materials, which suggests the kinetic stabilities of these functionalized MXenes. The M2CT2-I materials with functional groups located above the opposite-side metal atoms have the largest cohesive energies, and thus, are energetically most favorable. Significantly, we found that all the energetically favorable M2CO2-I materials have not only suitable band gaps (0.92–1.75 eV), but also visible-light absorption, high redox potential of photo-induced holes, efficient separation of e−–h+ pairs, and exceptional carrier mobilities, implying the great possibility of Ti2CO2, Zr2CO2 and Hf2CO2 materials to be utilized as visible-light driven photocatalysts. Additionally, the Hf2CO2 materials with appropriate band edges could be used for photocatalytic water splitting. Our theoretical studies are valuable for further exploring the potential applications of MXenes, which are worthy of experimental investigation and confirmations.
 Please wait while we load your content...
Please wait while we load your content...

