
Accurate calculation of the absolute free energy of binding for drug molecules†

Abstract
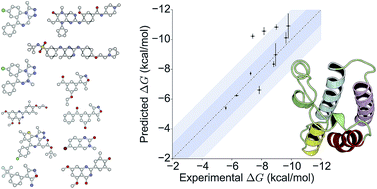
Accurate prediction of binding affinities has been a central goal of computational chemistry for decades, yet remains elusive. Despite good progress, the required accuracy for use in a drug-discovery context has not been consistently achieved for drug-like molecules. Here, we perform absolute free energy calculations based on a thermodynamic cycle for a set of diverse inhibitors binding to bromodomain-containing protein 4 (BRD4) and demonstrate that a mean absolute error of 0.6 kcal mol−1 can be achieved. We also show a similar level of accuracy (1.0 kcal mol−1) can be achieved in pseudo prospective approach. Bromodomains are epigenetic mark readers that recognize acetylation motifs and regulate gene transcription, and are currently being investigated as therapeutic targets for cancer and inflammation. The unprecedented accuracy offers the exciting prospect that the binding free energy of drug-like compounds can be predicted for pharmacologically relevant targets.
- This article is part of the themed collections: Top 50 Articles of 2016: Physical Chemistry, Top 50 Articles of 2016: Analytical, Biological and Medicinal Chemistry and Global challenges: Health & Food
 Please wait while we load your content...
Please wait while we load your content...

