
Reactive oxygen species in iridium-based OER catalysts†
 d
d
Abstract
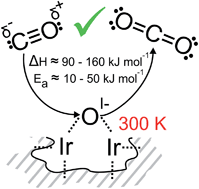
Tremendous effort has been devoted towards elucidating the fundamental reasons for the higher activity of hydrated amorphous IrIII/IV oxyhydroxides (IrOx) in the oxygen evolution reaction (OER) in comparison with their crystalline counterpart, rutile-type IrO2, by focusing on the metal oxidation state. Here we demonstrate that, through an analogy to photosystem II, the nature of this reactive species is not solely a property of the metal but is intimately tied to the electronic structure of oxygen. We use a combination of synchrotron-based X-ray photoemission and absorption spectroscopies, ab initio calculations, and microcalorimetry to show that holes in the O 2p states in amorphous IrOx give rise to a weakly bound oxygen that is extremely susceptible to nucleophilic attack, reacting stoichiometrically with CO already at room temperature. As such, we expect this species to play the critical role of the electrophilic oxygen involved in O–O bond formation in the electrocatalytic OER on IrOx. We propose that the dynamic nature of the Ir framework in amorphous IrOx imparts the flexibility in Ir oxidation state required for the formation of this active electrophilic oxygen.
 Please wait while we load your content...
Please wait while we load your content...

