
Precious-metal free photoelectrochemical water splitting with immobilised molecular Ni and Fe redox catalysts†
Abstract
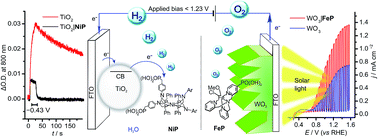
Splitting water into hydrogen and oxygen with molecular catalysts and light has been a long-established challenge. Approaches in homogeneous systems have been met with little success and the integration of molecular catalysts in photoelectrochemical cells is challenging due to inaccessibility and incompatibility of functional hybrid molecule/material electrodes with long-term stability in aqueous solution. Here, we present the first example of light-driven water splitting achieved with precious-metal-free molecular catalysts driving both oxygen and hydrogen evolution reactions. Mesoporous TiO2 was employed as a low-cost scaffold with long-term stability for anchoring a phosphonic acid-modified nickel(II) bis-diphosphine catalyst (NiP) for electrocatalytic proton reduction. A turnover number of 600 mol H2 per mol NiP was achieved after 8 h controlled-potential electrolysis at a modest overpotential of 250 mV. X-ray photoelectron, UV-vis and IR spectroscopies confirmed that the molecular structure of the Ni catalyst remains intact after prolonged hydrogen production, thereby reasserting the suitability of molecular catalysts in the development of effective, hydrogen-evolving materials. The relatively mild operating conditions of a pH 3 aqueous solution allowed this molecule-catalysed cathode to be combined with a molecular Fe(II) catalyst-modified WO3 photoanode in a photoelectrochemical cell. Water splitting into H2 and O2 was achieved under solar light illumination with an applied bias of >0.6 V, which is below the thermodynamic potential (1.23 V) for water splitting and therefore allowed the storage of solar energy in the fuel H2.
- This article is part of the themed collections: In celebration of Kazunari Domen’s 65th birthday, 2018, ISACS17: Challenges in Chemical Renewable Energy, Global Energy Challenges: Solar Energy and Global Energy Challenges: Hydrogen Energy
 Please wait while we load your content...
Please wait while we load your content...

