
Carbon rings: a DFT study on geometry, aromaticity, intermolecular carbon–carbon interactions and stability†
Abstract
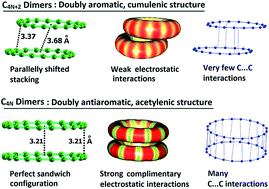
Non-covalent dimer formation and intermolecular bonding features of planar monocyclic carbon rings showing C4N+2 and C4N configurations have been studied using the meta-GGA DFT method, M06L/6-311+G(d) for N = 1–8. The C4N+2 show cumulenic structures with equal bond lengths and C4N form structures with clear bond length alternation. The doubly Hückel aromatic nature of C4N+2 is revealed through two cyclic delocalized π-molecular orbitals and highly negative nucleus independent chemical shift (NICS) parameters while the doubly Hückel antiaromatic nature of C4N is brought out through two localized π-molecular orbitals and highly positive NICS parameters. Further, the uniform electron distribution over the delocalized CC bonds in C4N+2 and the alternate electron rich and electron deficient regions in C4N are assessed on the basis of the critical features of the molecular electrostatic potential (MESP). The contrasting geometric, electronic and magnetic features of C4N+2 compared to C4N result in a drastic difference in their intermolecular bonding behaviour. The C4N showed a much higher tendency than C4N+2 for dimer formation as the former, in general show a 4N number of intermolecular C⋯C interactions due to complimentary electrostatic interactions between electron rich shorter CC bonds and electron deficient longer CC bonds. In C4N dimers, a perfect sandwich configuration is preferred to maximize the attractive complementary electrostatic interactions while in C4N+2 dimers a shifted-parallel stacked arrangement indicated the non-complementary character of interactions arising from smooth aromatic distribution of electrons. The comparative stability of the carbon rings and unsubstituted polyynes is quantified by measuring the homodesmotic reaction energy (Ehdr) with acetylene. The Ehdr indicated significant stabilization of C4N+2 compared to C4N. The energy required to open up a carbon ring to the linear form is computed as Eopening and this quantity is used to estimate the aromatic stabilization of C4N+2 as well as the antiaromatic destabilization of C4N systems.
 Please wait while we load your content...
Please wait while we load your content...

