
Cation–π interactions in CREBBP bromodomain inhibition: an electrostatic model for small-molecule binding affinity and selectivity†

Abstract
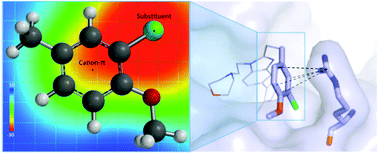
CREBBP bromodomains, epigenetic “reader” proteins that recognize acetylated histone lysine residues, are a current target for cancer therapy. We show that experimental CREBBP binding affinities of small-molecules with aromatic or heteroaromatic functional groups are strongly influenced by a cation–π interaction with a positively charged arginine residue. For a series of fifteen 5-isoxazolylbenzimidazole derivatives, the strength of this non-covalent interaction is directly related to improvements in binding to CREBBP. The aromatic substituents’ inductive and resonance effects are not obviously correlated with observed structure and affinity relationships. In contrast, a coulombic electrostatic model can quantitatively predict the interaction strength. We have assessed different Molecular Mechanics (MM) and Quantum Mechanics (QM) descriptions of the protein–ligand interaction. Quantitative models for binding affinity were generated from: (1) Poisson Boltzmann Surface Area (MM-PBSA) and Generalized Born Surface Area (MM-GBSA) scoring functions that incorporated the entire ligand and (2) QM-complexation energies and (3) Electrostatic Potential Surface values (ESPs) that analyzed the varying aromatic group. A linear relationship between QM-computed ESP values is established for the cation–π interaction strength, and gives the best correlation (R2 = 0.84) with experimental binding affinities. This model also ranks ligand affinity most accurately (rs = 0.91) from the models tested. Consideration of the electrostatic potential in response to the local effects of substituents in addition to that of the aromatic ring is necessary to understand and describe the interaction with the cationic guanidinium ion. This leads to an improved understanding and the ability to quantitatively predict the magnitude of non-covalent interactions in the CREBBP active site.
 Please wait while we load your content...
Please wait while we load your content...

