
Asymmetric synthesis of crambescin A–C carboxylic acids and their inhibitory activity on voltage-gated sodium channels†‡
Abstract
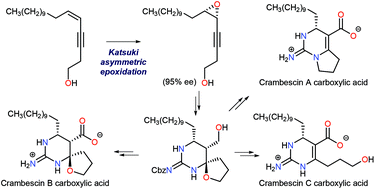
Synthesis of both enantiomers of crambescin B carboxylic acid is described. A cis-enyne starting material was epoxidized under the conditions of Katsuki asymmetric epoxidation to give 95% ee of the epoxide, which was transformed to crambescin B carboxylic acid via bromocation-triggered cascade cyclization as the key step. Enantiomerically pure crambescin A and C carboxylic acids were also synthesized from the product of the cascade reaction. Structure–activity relationship (SAR) studies against voltage-gated sodium channel (VGSC) inhibition using those synthetic compounds revealed that the natural enantiomer of crambescin B carboxylic acid was most active and comparable to tetrodotoxin, and the unalkylated cyclic guanidinium structure is indispensible, while the carboxylate moiety is not important. The absolute stereochemistry of crambescin A was determined by a comparison of the methyl ester derived from natural crambescin A with that derived from the stereochemically defined crambescin A carboxylic acid synthesized in this study.
 Please wait while we load your content...
Please wait while we load your content...

