
The kinetics and mechanism of the organo-iridium-catalysed enantioselective reduction of imines†
Abstract
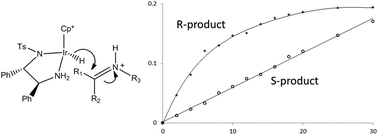
The iridium complex of pentamethylcyclopentadiene and (S,S)-1,2-diphenyl-N′-tosylethane-1,2-diamine is an effective catalyst for the asymmetric transfer hydrogenation of imine substrates under acidic conditions. Using the Ir catalyst and a 5 : 2 ratio of formic acid : triethylamine as the hydride source for the asymmetric transfer hydrogenation of 1-methyl-3,4-dihydroisoquinoline and its 6,7-dimethoxy substituted derivative, in either acetonitrile or dichloromethane, shows unusual enantiomeric excess (ee) profiles for the product amines. The reactions initially give predominantly the (R) enantiomer of the chiral amine products with >90% ee but which then decreases significantly during the reaction. The decrease in ee is not due to racemisation of the product amine, but because the rate of formation of the (R)-enantiomer follows first-order kinetics whereas that for the (S)-enantiomer is zero-order. This difference in reaction order explains the change in selectivity as the reaction proceeds – the rate formation of the (R)-enantiomer decreases exponentially with time while that for the (S)-enantiomer remains constant. A reaction scheme is proposed which requires rate-limiting hydride transfer from the iridium hydride to the iminium ion for the first-order rate of formation of the (R)-enantiomer amine and rate-limiting dissociation of the product for the zero-order rate of formation of the (S)-enantiomer.
 Please wait while we load your content...
Please wait while we load your content...

