
Computer-aided molecular design and selection of CO2 capture solvents based on thermodynamics, reactivity and sustainability†
 c
and
c
and
Abstract
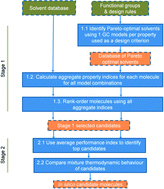
The identification of improved carbon dioxide (CO2) capture solvents remains a challenge due to the vast number of potentially-suitable molecules. We propose an optimization-based computer-aided molecular design (CAMD) method to identify and select, from hundreds of thousands of possibilities, a few solvents of optimum performance for CO2 chemisorption processes, as measured by a comprehensive set of criteria. The first stage of the approach involves a fast screening stage where solvent structures are evaluated based on the simultaneous consideration of important pure component properties reflecting thermodynamic, kinetic, and sustainability behaviour. The impact of model uncertainty is considered through a systematic method that employs multiple models for the prediction of performance indices. In the second stage, high-performance solvents are further selected and evaluated using a more detailed thermodynamic model, i.e. the group-contribution statistical associating fluid theory for square well potentials (SAFT-γ SW), to predict accurately the highly non-ideal chemical and phase equilibrium of the solvent–water–CO2 mixtures. The proposed CAMD method is applied to the design of novel molecular structures and to the screening of a data set of commercially available amines. New molecular structures and commercially-available compounds that have received little attention as CO2 capture solvents are successfully identified and assessed using the proposed approach. We recommend that these solvents should be given priority in experimental studies to identify new compounds.
 Please wait while we load your content...
Please wait while we load your content...

