
Abstract
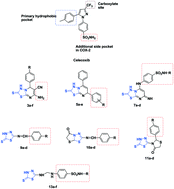
A novel series of thiadiazole derivatives were designed and synthesized for evaluation as selective COX-2 inhibitors in vitro and were investigated in vivo as anti-inflammatory and analgesic agents against carrageenan-induced rat paw oedema model in irradiated rats, since it is well-known that ionizing radiation plays an important role in exaggerating the inflammatory responses in experimental animals. Ulcerogenic activity and acute toxicity were evaluated for the most potent compounds. In order to understand the binding mode of the synthesized compounds into the active site of the COX-2 enzyme, a docking study was performed. Most of the tested compounds showed high inhibitory ability against COX-2. Among them, thiadiazole derivatives bearing sulfonamide moieties, 7b, c and 13c, e, were the most potent COX-2 inhibitors, with extremely high selectivity indices (SI) compared to celecoxib; they showed high safety margins on gastric mucosa with no ulceration effects, and they were found to be non-toxic in experimental rats. The docking study showed a similar orientation of these compounds to that of celecoxib within the active site of the COX-2 enzyme, and a similar ability to immerge deeply into the additional pocket and bind with Arg513 and His90, the key amino acids responsible for selectivity.
 Please wait while we load your content...
Please wait while we load your content...