
Molecular insight on the binding of NNRTI to K103N mutated HIV-1 RT: molecular dynamics simulations and dynamic pharmacophore analysis†
 a
a
Abstract
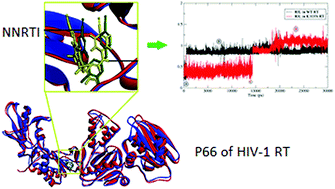
Regardless of advances in anti-HIV therapy, HIV infection remains an immense challenge due to the rapid onset of mutation instigating drug resistance. Rilpivirine is a second generation di-aryl pyrimidine (DAPY) derivative, known to effectively inhibit wild-type (WT) as well as various mutant HIV-1 reverse transcriptase (RT). In this study, a cumulative 240 ns of molecular dynamic (MD) simulations of WT HIV-1 RT and its corresponding K103N mutated form, complexed with rilpivirine, were performed in solution. Conformational analysis of the NNRTI inside the binding pocket (NNIBP) revealed the ability of rilpivirine to adopt different conformations, which is possibly the reason for its reasonable activity against mutant HIV-1 RT. Binding free energy (MM-PB/GB SA) calculations of rilpivirine with mutant HIV-1 RT are in agreement with experimental data. The dynamics of interaction patterns were investigated based on the MD simulations using dynophores, a novel approach for MD-based ligand–target interaction mapping. The results from this interaction profile analysis suggest an alternate interaction between the linker N atom of rilpivirine and Lys 101, potentially providing the stability for ligand binding. PCA analysis and per residue fluctuation has highlighted the significant role of flexible thumb and finger sub-domains of RT in its biological activity. This study investigated the underlying reason for rilpivirine's improved inhibitory profile against mutant RT, which could be helpful to understand the molecular basis of HIV-1 RT drug resistance and design novel NNRTIs with improved drug resistance tolerance.
 Please wait while we load your content...
Please wait while we load your content...