
Electronic communication in phosphine substituted bridged dirhenium complexes – clarifying ambiguities raised by the redox non-innocence of the C4H2- and C4-bridges†
Abstract
The mononuclear rhenium carbyne complex trans-[Re(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CSiMe3)(C–Me)(PMe3)4][PF6] (2) was prepared in 90% yield by heating a mixture of the dinitrogen complex trans-[ReCl(N2)(PMe3)4] (1), TlPF6, and an excess of HCCSiMe3. 2 could be deprotonated with KOtBu to the vinylidene complex trans-[Re(CCSiMe3)(
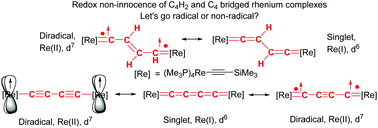
CSiMe3)(C–Me)(PMe3)4][PF6] (2) was prepared in 90% yield by heating a mixture of the dinitrogen complex trans-[ReCl(N2)(PMe3)4] (1), TlPF6, and an excess of HCCSiMe3. 2 could be deprotonated with KOtBu to the vinylidene complex trans-[Re(CCSiMe3)(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CCH2)(PMe3)4] (3) in 98% yield. Oxidation of 3 with 1.2 equiv. of [Cp2Fe][PF6] at −78 °C gave the Cβ–C′β coupled dinuclear rhenium biscarbyne complex trans-[(Me3SiCC)(PMe3)4ReC–CH2–CH2–CRe(PMe3)4(CCSiMe3)][PF6]2 (5) in 92% yield. Deprotonation of 5 with an excess of KOtBu in THF produced the diamagnetic trans-[(Me3SiCC)(PMe3)4ReCCH–CHCRe(PMe3)4(CCSiMe3)] complex (E-6(S)) in 87% yield with an E-butadienediylidene bridge. Density functional theory (DFT) calculations of E-6(S) confirmed its singlet ground state. The Z-form of 6 (Z-6(S)) could not be observed, which is in accord with its DFT calculated 17.8 kJ mol−1 higher energy. Oxidation of E-6 with 2 equiv. of [Cp2Fe][PF6] resulted in the stable diamagnetic dicationic trans-[(Me3SiCC)(PMe3)4ReC–CHCH–CRe(PMe3)4(CCSiMe3)][PF6]2 complex (E-6[PF6]2) with an ethylenylidene dicarbyne structure of the bridge. The paramagnetic mixed-valence (MV) complex E-6[PF6] was obtained by comproportionation of E-6(S) and E-6[PF6]2 or by oxidation of E-6(S) with 1 equiv. of [Cp2Fe][PF6]. The dicationic trans-[(Me3SiCC)(PMe3)4ReC–CC–CRe(PMe3)4(CCSiMe3)][PF6]2 (7[PF6]2) complex, attributed a butynedi(triyl) bridge structure, was obtained by deprotonation of E-6[PF6]2 with KOtBu followed by oxidation with 2 equiv. of [Cp2Fe][PF6]. The neutral complex 7 could be accessed best by reduction of 7[PF6]2 with KH in the presence of 18-crown-6. According to DFT calculations 7 possesses two equilibrating electronic states: diamagnetic 7(S) and triplet 7(F) with ferromagnetically coupled spins. The latter is calculated to be 5.2 kcal mol−1 lower in energy than 7(S). There is experimental evidence that 7(S) prevails in solution. 7 could not be isolated in the crystalline state and is unstable transforming mainly by H-abstraction to give E-6(S). UV-Vis-NIR spectroscopy for the dinuclear rhenium complexes E-6(S), E-6[PF6] and E-6[PF6]2, as well as EPR spectroscopic and variable-temperature magnetization measurements for the MV complex E-6[PF6] were also conducted. Spectro-electrochemical reduction studies on 7[PF6]2 allowed the characterization of the mono- and direduced forms of 7+ and 7 by means of IR- and UV-Vis-NIR-spectroscopy and revealed the chemical fate of the higher reduced form.
CCH2)(PMe3)4] (3) in 98% yield. Oxidation of 3 with 1.2 equiv. of [Cp2Fe][PF6] at −78 °C gave the Cβ–C′β coupled dinuclear rhenium biscarbyne complex trans-[(Me3SiCC)(PMe3)4ReC–CH2–CH2–CRe(PMe3)4(CCSiMe3)][PF6]2 (5) in 92% yield. Deprotonation of 5 with an excess of KOtBu in THF produced the diamagnetic trans-[(Me3SiCC)(PMe3)4ReCCH–CHCRe(PMe3)4(CCSiMe3)] complex (E-6(S)) in 87% yield with an E-butadienediylidene bridge. Density functional theory (DFT) calculations of E-6(S) confirmed its singlet ground state. The Z-form of 6 (Z-6(S)) could not be observed, which is in accord with its DFT calculated 17.8 kJ mol−1 higher energy. Oxidation of E-6 with 2 equiv. of [Cp2Fe][PF6] resulted in the stable diamagnetic dicationic trans-[(Me3SiCC)(PMe3)4ReC–CHCH–CRe(PMe3)4(CCSiMe3)][PF6]2 complex (E-6[PF6]2) with an ethylenylidene dicarbyne structure of the bridge. The paramagnetic mixed-valence (MV) complex E-6[PF6] was obtained by comproportionation of E-6(S) and E-6[PF6]2 or by oxidation of E-6(S) with 1 equiv. of [Cp2Fe][PF6]. The dicationic trans-[(Me3SiCC)(PMe3)4ReC–CC–CRe(PMe3)4(CCSiMe3)][PF6]2 (7[PF6]2) complex, attributed a butynedi(triyl) bridge structure, was obtained by deprotonation of E-6[PF6]2 with KOtBu followed by oxidation with 2 equiv. of [Cp2Fe][PF6]. The neutral complex 7 could be accessed best by reduction of 7[PF6]2 with KH in the presence of 18-crown-6. According to DFT calculations 7 possesses two equilibrating electronic states: diamagnetic 7(S) and triplet 7(F) with ferromagnetically coupled spins. The latter is calculated to be 5.2 kcal mol−1 lower in energy than 7(S). There is experimental evidence that 7(S) prevails in solution. 7 could not be isolated in the crystalline state and is unstable transforming mainly by H-abstraction to give E-6(S). UV-Vis-NIR spectroscopy for the dinuclear rhenium complexes E-6(S), E-6[PF6] and E-6[PF6]2, as well as EPR spectroscopic and variable-temperature magnetization measurements for the MV complex E-6[PF6] were also conducted. Spectro-electrochemical reduction studies on 7[PF6]2 allowed the characterization of the mono- and direduced forms of 7+ and 7 by means of IR- and UV-Vis-NIR-spectroscopy and revealed the chemical fate of the higher reduced form.
 Please wait while we load your content...
Please wait while we load your content...

