
Computational investigation into the gas-phase ozonolysis of the conjugated monoterpene α-phellandrene†
Abstract
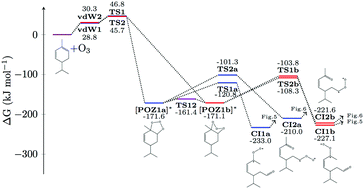
Reaction with ozone is a major atmospheric sink for α-phellandrene, a monoterpene found in both indoor and outdoor environments, however experimental literature concerning the reaction is scarce. In this study, high-level G4(MP2) quantum chemical calculations are used to theoretically characterise the reaction of ozone with both double bonds in α-phellandrene for the first time. Results show that addition of ozone to the least substituted double bond in the conjugated system is preferred. Following addition, thermal and chemically activated unimolecular reactions, including the so-called hydroperoxide and ester or ‘hot’ acid channels, and internal cyclisation reactions, are characterised to major first generation products. Conjugation present in α-phellandrene allows two favourable Criegee intermediate reaction pathways to proceed that have not previously been considered in the literature; namely a 1,6-allyl resonance stabilised hydrogen shift and intramolecular dioxirane isomerisation to an epoxide. These channels are expected to play an important role alongside conventional routes in the ozonolysis of a-phellandrene. Computational characterisation of the potential energy surface thus provides insight into this previously unstudied system, and will aid future mechanism development and experimental interpretation involving α-phellandrene and structurally similar species, to which the results are expected to extend.
 Please wait while we load your content...
Please wait while we load your content...

