
Molecular dynamics simulations of mixtures of protic and aprotic ionic liquids
Abstract
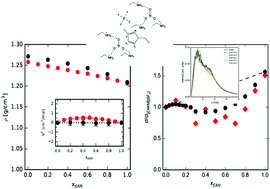
Molecular dynamics simulations of mixtures of the protic ionic liquid ethylammonium nitrate (EAN) and the aprotic 1-ethyl-3-methylimidazolium tetrafluoroborate ([EMIM][BF4]) are reported and the results are compared with experimental density and electrical conductivity measurements. Essentially ideal mixing of the ionic liquids is seen to take place by means of experimental and simulated excess molar volumes, whose very low values suggest a gradual transition between the structures of the two end constituents of the mixture. A weak dominance of the structure of the protic ionic liquid is nevertheless registered, due to a slight preferential formation of the network of hydrogen bonds, as reflected in the coordination number and the number of hydrogen bonds in the mixture. A novel conductivity curve showing pronounced deviations from the simple ideal mixing rule is reported, with three different regions defined by a local maximum – reflecting enhanced translational dynamics relative to ideal mixture behaviour – and a global minimum at intermediate concentrations. The physical origin of this behaviour is discussed along with the structure and single-particle dynamics of the mixture, and it is seen that these regions are defined by the onset of the formation of the EAN hydrogen bonded network (xEAN = 0.2) and the virtual disappearance of the structure of the aprotic ionic liquid at xEAN = 0.7. It is concluded that the delicate interplay between both networks has a deep effect on the placement and mobility of [EMIM]+ cations in the mixture all throughout the different stages of the structural transition, which seems to be the driving force behind the reported transport properties of the mixture at intermediate to high EAN concentrations.
 Please wait while we load your content...
Please wait while we load your content...

