
Cyclotetrahalo-p-phenylenes: simulations of halogen substituted cycloparaphenylenes and their interaction with C60†
 a
a
Abstract
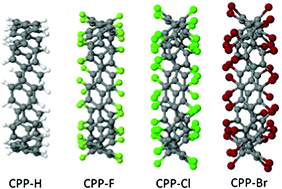
Density functional calculations are used to study the role of edge-functionalization on the structure and electronic properties of cycloparaphenylene (CPPs) containing from six to twenty benzenoid rings. We substitute hydrogen by the halogens fluorine, chlorine and bromine. The resultant Cyclotetrahalo-p-phenylenes are compared with their hydrogenated equivalents, related linear paraphenyl and fluoro-paraphenyl polymers, and functionalised armchair edges in graphene nanoribbons. Notably we consider both structural and electronic evolution. Finally we examine C60@[10]CPP, i.e. C60 encapsulated within [10]CPP, with the various ring terminations. The effect of halogenation on electronic level position around the gap strongly affects their capacity to form donor–acceptor pairs with fullerenes.
 Please wait while we load your content...
Please wait while we load your content...

