
Density functional simulation of resonant inelastic X-ray scattering experiments in liquids: acetonitrile†
Abstract
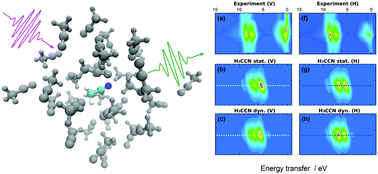
In this paper we report an experimental and computational study of liquid acetonitrile (H3C–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N) by resonant inelastic X-ray scattering (RIXS) at the N K-edge. The experimental spectra exhibit clear signatures of the electronic structure of the valence states at the N site and incident-beam-polarization dependence is observed as well. Moreover, we find fine structure in the quasielastic line that is assigned to finite scattering duration and nuclear relaxation. We present a simple and light-to-evaluate model for the RIXS maps and analyze the experimental data using this model combined with ab initio molecular dynamics simulations. In addition to polarization-dependence and scattering-duration effects, we pinpoint the effects of different types of chemical bonding to the RIXS spectrum and conclude that the H2C–C
N) by resonant inelastic X-ray scattering (RIXS) at the N K-edge. The experimental spectra exhibit clear signatures of the electronic structure of the valence states at the N site and incident-beam-polarization dependence is observed as well. Moreover, we find fine structure in the quasielastic line that is assigned to finite scattering duration and nuclear relaxation. We present a simple and light-to-evaluate model for the RIXS maps and analyze the experimental data using this model combined with ab initio molecular dynamics simulations. In addition to polarization-dependence and scattering-duration effects, we pinpoint the effects of different types of chemical bonding to the RIXS spectrum and conclude that the H2C–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NH isomer, suggested in the literature, does not exist in detectable quantities. We study solution effects on the scattering spectra with simulations in liquid and in vacuum. The presented model for RIXS proved to be light enough to allow phase-space-sampling and still accurate enough for identification of transition lines in physical chemistry research by RIXS.
NH isomer, suggested in the literature, does not exist in detectable quantities. We study solution effects on the scattering spectra with simulations in liquid and in vacuum. The presented model for RIXS proved to be light enough to allow phase-space-sampling and still accurate enough for identification of transition lines in physical chemistry research by RIXS.
 Please wait while we load your content...
Please wait while we load your content...

