
Rovibrational energy levels of the F−(H2O) and F−(D2O) complexes†

Abstract
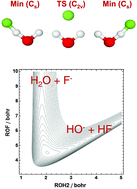
The variational nuclear-motion codes ElVibRot and GENIUSH have been used to compute rotational–vibrational states of the F−(H2O) anion and its deuterated isotopologue, F−(D2O), employing a full-dimensional, semiglobal potential energy surface (PES) called SLBCL, developed as part of this study for the ground electronic state of the complex. The PES is determined from all-electron, explicitly correlated coupled-cluster singles, doubles, and connected triples [CCSD(T)-F12a] computations with an atom-centered, fixed-exponent Gaussian basis set of cc-pCVTZ-F12 quality. The SLBCL PES accurately reproduces the two equivalent minima of the complex, the corresponding transition barrier of C2v point-group symmetry, as well as the proton transfer and the dissociation asymptotes towards the products HF + OH− and F− + H2O, respectively. The code ElVibRot has been updated so that it can use curvilinear internal coordinates corresponding to a reaction path. The variationally computed vibrational energy levels are compared to relevant experimental and previously determined first-principles results. The vibrational states reveal the presence of pronounced anharmonic effects and considerable intermode couplings resulting in strong resonances, involving in particular the HOH bend and the ionic OH stretch motions. Tunneling results in particularly significant splittings for F−(H2O); as expected, the splittings are orders of magnitude smaller for the F−(D2O) molecule. The rovibrational energy levels reveal that, despite the large-amplitude vibrational motions, the rotations of F−(H2O) basically follow rigid-rotor characteristics.
 Please wait while we load your content...
Please wait while we load your content...

