
Electronic origin of the dependence of hydrogen bond strengths on nearest-neighbor and next-nearest-neighbor hydrogen bonds in polyhedral water clusters (H2O)n, n = 8, 20 and 24†
Abstract
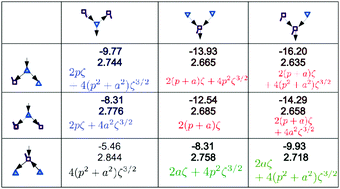
The influence of the nearest neighbor and next-nearest neighbor water molecules on the strength of the hydrogen (H) bonds was examined for the polyhedral clusters of cubic (H2O)8, dodecahedral (H2O)20 and tetrakaidecahedral (H2O)24 cages. The relative stability and the characteristics of the H bond networks are also studied. The charge-transfer (CT) and dispersion interaction terms of every pair of H bonds are evaluated using perturbation theory based on the locally-projected molecular orbitals (LPMO PT). Every water molecule and every H-bonded pair in these polyhedral clusters are classified by the types of the neighbor molecules and H bonds. The relative binding energies among the polyhedral clusters are grouped by these classifications. The optimized O⋯O distances, which are strongly correlated with the calculated pairwise CT terms, are dependent on the 49 sub-groups of the H bonds determined by the type of the neighbor molecules. The electronic origin of this dependence is analyzed using Mulliken's charge-transfer theory, and employing a few assumptions, the analytical formulas for the contribution of the CT terms to the H bond energy are derived.
 Please wait while we load your content...
Please wait while we load your content...

