
Screened exchange hybrid density functional for accurate and efficient structures and interaction energies†
Abstract
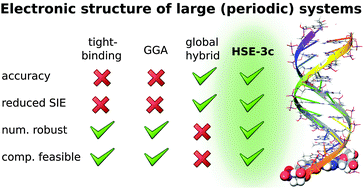
We extend the recently introduced PBEh-3c global hybrid density functional [S. Grimme et al., J. Chem. Phys., 2015, 143, 054107] by a screened Fock exchange variant based on the Henderson–Janesko–Scuseria exchange hole model. While the excellent performance of the global hybrid is maintained for small covalently bound molecules, its performance for computed condensed phase mass densities is further improved. Most importantly, a speed up of 30 to 50% can be achieved and especially for small orbital energy gap cases, the method is numerically much more robust. The latter point is important for many applications, e.g., for metal–organic frameworks, organic semiconductors, or protein structures. This enables an accurate density functional based electronic structure calculation of a full DNA helix structure on a single core desktop computer which is presented as an example in addition to comprehensive benchmark results.
 Please wait while we load your content...
Please wait while we load your content...

