
The S66x8 benchmark for noncovalent interactions revisited: explicitly correlated ab initio methods and density functional theory†
 b
and
b
and
Abstract
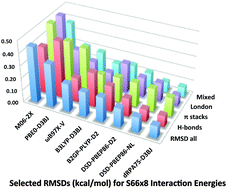
The S66x8 dataset for noncovalent interactions of biochemical relevance has been re-examined by means of MP2-F12 and CCSD(F12*)(T) methods. We deem our revised benchmark data to be reliable to about 0.05 kcal mol−1 RMS. Most levels of DFT perform quite poorly in the absence of dispersion corrections: somewhat surprisingly, that is even the case for the double hybrids and for dRPA75. Analysis of optimized D3BJ parameters reveals that the main benefit of dRPA75 and DSD double hybrids alike is the treatment of midrange dispersion. dRPA75-D3BJ is the best performer overall at RMSD = 0.10 kcal mol−1. The nonlocal VV10 dispersion functional is especially beneficial for the double hybrids, particularly in DSD-PBEP86-NL (RMSD = 0.12 kcal mol−1). Other recommended dispersion-corrected functionals with favorable price/performance ratios are ωB97X-V, and, surprisingly, B3LYP-D3BJ and BLYP-D3BJ (RMSDs of 0.23, 0.20 and 0.23 kcal mol−1, respectively). Without dispersion correction (but parametrized for midrange interactions) M06-2X has the lead (RMSD = 0.45 kcal mol−1). A collection of three energy-based diagnostics yields similar information to an SAPT analysis about the nature of the noncovalent interaction. Two of those are the percentages of Hartree–Fock and of post-MP2 correlation effects in the interaction energy; the third, CSPI = [IE(2)ss − IE(2)ab]/[IE(2)ss + IE(2)ab] or its derived quantity DEBC = CSPI/(1 + CSPI2)1/2, describes the character of the MP2 correlation contribution, ranging from 0 (purely dispersion) to 1 (purely other effects). In addition, we propose an improved, parameter-free scaling for the (T) contribution based on the Ecorr[CCSD-F12b]/Ecorr[CCSD] and Ecorr[CCSD(F12*)]/Ecorr[CCSD] ratios. For Hartree–Fock and conventional DFT calculations, full counterpoise generally yields the fastest basis set convergence, while for double hybrids, half-counterpoise yields faster convergence, as previously established for correlated ab initio methods.
- This article is part of the themed collection: Developments in Density Functional Theory
 Please wait while we load your content...
Please wait while we load your content...

