
NO adsorption on Cu(110) and O(2 × 1)/Cu(110) surfaces from density functional theory calculations
Abstract
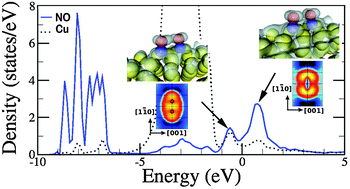
In a recent study [M. Feng, et al., ACS Nano, 2011, 5, 8877], it was shown that CO molecules adsorbed on the quasi-one-dimensional O(2 × 1)/Cu(110) surface reconstruction tend to form highly-ordered single-molecule-wide rows along the direction perpendicular to the Cu–O chains. This stems from the peculiar tilted adsorption configuration of CO on this substrate, which gives rise to short-range attractive dipole–dipole interactions. Motivated by this observation, here we study the adsorption of nitric oxide (NO) on O(2 × 1)/Cu(110) and Cu(110) using density functional theory, with the aim of elucidating whether a similar behaviour can be expected for this molecule. We first study NO adsorption on a clean Cu(110) surface, where the role of short-range attractions between molecules has already been pointed out by the observation of the formation of NO dimers by scanning tunnelling microscopy [A. Shiotari, et al., Phys. Rev. Lett., 2011, 106, 156104]. On the clean Cu(110), the formation of dimers along the [1![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 0] direction is favourable, in agreement with published experimental results. However, the formation of extended NO rows is found to be unstable. Regarding the O(2 × 1)/Cu(110) substrate, we observe that NO molecules adsorb in between the Cu–O chains, causing a substantial disruption of the surface structure. Although individual molecules can be tilted with negligible energetic cost along the direction of the Cu–O chains, the interaction among neighbouring molecules was found to be repulsive along all directions and, consequently, the formation of dimers unfavourable.
0] direction is favourable, in agreement with published experimental results. However, the formation of extended NO rows is found to be unstable. Regarding the O(2 × 1)/Cu(110) substrate, we observe that NO molecules adsorb in between the Cu–O chains, causing a substantial disruption of the surface structure. Although individual molecules can be tilted with negligible energetic cost along the direction of the Cu–O chains, the interaction among neighbouring molecules was found to be repulsive along all directions and, consequently, the formation of dimers unfavourable.
 Please wait while we load your content...
Please wait while we load your content...

