
Computing UV/vis spectra using a combined molecular dynamics and quantum chemistry approach: bis-triazin-pyridine (BTP) ligands studied in solution†
Abstract
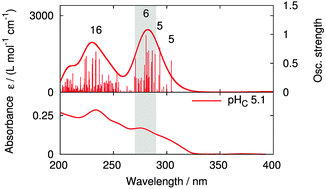
We report a combined computational and experimental study to investigate the UV/vis spectra of 2,6-bis(5,6-dialkyl-1,2,4-triazin-3-yl)pyridine (BTP) ligands in solution. In order to study molecules in solution using theoretical methods, force-field parameters for the ligand–water interaction are adjusted to ab initio quantum chemical calculations. Based on these parameters, molecular dynamics (MD) simulations are carried out from which snapshots are extracted as input to quantum chemical excitation-energy calculations to obtain UV/vis spectra of BTP ligands in solution using time-dependent density functional theory (TDDFT) employing the Tamm–Dancoff approximation (TDA). The range-separated CAM-B3LYP functional is used to avoid large errors for charge-transfer states occurring in the electronic spectra. In order to study environment effects with theoretical methods, the frozen-density embedding scheme is applied. This computational procedure allows to obtain electronic spectra calculated at the (range-separated) DFT level of theory in solution, revealing solvatochromic shifts upon solvation of up to about 0.6 eV. Comparison to experimental data shows a significantly improved agreement compared to vacuum calculations and enables the analysis of relevant excitations for the line shape in solution.
 Please wait while we load your content...
Please wait while we load your content...

