
Ammonia-modified Co(ii) sites in zeolites: spin and electron density redistribution through the CoII–NO bond†
Abstract
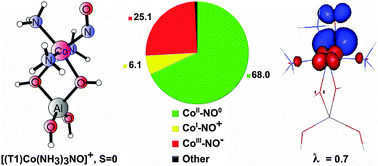
Electronic factors essential for the bonding of a non-innocent NO ligand to ammonia-modified Co2+ sites in cobalt-exchanged zeolites are examined for small cluster models using DFT and advanced correlated wave function calculations. The analysis of charge transfer processes between the NO ligand and the cobalt center involves two protocols: valence-bond expansion of the multiconfiguration CASSCF wave function (in terms of fragment-localized active orbitals) and spin-resolved natural orbitals for chemical valence (SR-NOCV). Applicability of SR-NOCV analysis to transition metal complexes involving non-innocent fragments is critically assessed and the approach based on the CASSCF wave function turns out to be much more robust and systematic for all studied models. It is shown that the character and direction of electron density redistribution through the Co–N–O bond, quantified by relative share of the CoII–NO0, CoIII–NO−, and CoI–NO+ resonance structures in the total wave function, fully rationalize the activation of the N–O bond upon NH3 co-ligation (evidenced by calculated and measured red-shift of the NO stretching frequency and commonly ascribed to enhanced backdonation). The huge red-shift of νN–O is attributed to an effective electron transfer between the ammonia-modified Co(II) centers and the NO antibonding π*-orbitals (related to the increased share of the CoIII–NO− form). Unexpectedly, the effect is stronger for the singlet complex with three NH3 ligands than for that with five NH3 ligands bound to the cobalt center. Our results also indicate that high-efficiency electron transfers between the Co(II) center and the NO ligand may be enabled for the selected spin state and disabled for the other spin state of the adduct. This illustrates how the cobalt center may serve to fine-tune the electronic communication between the NO ligand and its binding site.
 Please wait while we load your content...
Please wait while we load your content...

