
Molecular structure and vibrations of NTCDA monolayers on Ag(111) from density-functional theory and infrared absorption spectroscopy†
Abstract
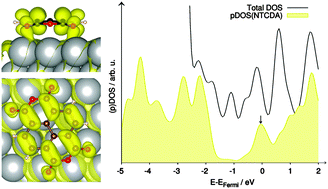
The structure and vibrational properties of the metal–organic interface of 1,4,5,8-naphthalene-tetracarboxylic dianhydride (NTCDA) on Ag(111) were analysed using Fourier-transform infrared absorption spectroscopy in conjunction with density functional theory calculations including dispersion forces (PBE-D3). Mode assignments and polarizations as well as molecular distortions were determined for four adsorption geometries of NTCDA on top and bridge sites aligned either parallel or perpendicular to the Ag rows and compared to accurate calculations of the free molecule. This enables an in-depth understanding of surface effects on the computed and experimental vibrational spectra of the adsorbed NTCDA molecule. The molecule–substrate interaction comprises two major and equally important contributions: non-directional van der Waals forces between molecule and surface, and covalent bonding of the acyl oxygen atoms with underlying Ag atoms, which is quantified by charge-transfer analysis. Furthermore, adsorption energy calculations showed that the molecular axis of flat-lying NTCDA is oriented preferably in parallel to the Ag rows. The molecule is subject to particular distortions from the planar gas phase structure with covalent bonding leading to downward bending of the acyl oxygen atoms and Pauli repulsion to upward bending of the carbon core. In parallel, strong buckling of the silver surface was identified. As found in previous studies, the lowest unoccupied molecular orbital (LUMO) of the molecule slips below the Fermi level and becomes partially populated upon adsorption. Excitation of totally symmetric vibrational modes then leads to substantial interfacial dynamical charge transfer, which is convincingly reproduced in the calculated IR spectra.
 Please wait while we load your content...
Please wait while we load your content...

