
Molecular simulations of imidazolium-based tricyanomethanide ionic liquids using an optimized classical force field†
Abstract
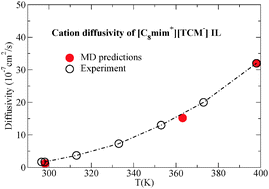
Imidazolium-based ionic liquids (ILs) incorporating the tricyanomethanide ([TCM−]) anion are studied using an optimized classical force field. These ILs are very promising candidates for use in a wide range of cutting-edge technologies and, to our knowledge, it is the first time that this IL family is subject to a molecular simulation study with the use of a classical atomistic force field. The [C4mim+][TCM−] ionic liquid at 298.15 K and at atmospheric pressure was used as the basis for force field optimization which primarily involved the determination of the Lennard-Jones parameters of [TCM-] and the implementation of three quantum mechanical schemes for the calculation of the partial charge distribution and the identification of the appropriate scaling factor for the reduction of the total ionic charge. The optimized force field was validated by performing simulations of the 1-alkyl-3-methylimidazolium tricyanomethanide ([Cnmim+][TCM−], n = 2, 4, 6, and 8) IL family at various temperatures. The results for density, self-diffusivity and viscosity are in very good agreement with the available experimental data for all ILs verifying that the force field reliably reproduces the behaviour of the imidazolium-based [TCM−] IL family in a wide temperature range. Furthermore, a detailed analysis of the microscopic structure and the complex dynamic behaviour of the ILs under study was performed.
 Please wait while we load your content...
Please wait while we load your content...

