
A graphene–boron nitride lateral heterostructure – a first-principles study of its growth, electronic properties, and chemical topology†
Abstract
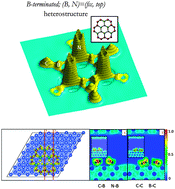
The lateral integration of graphene and hexagonal boron nitride permits the intricate design of a hybrid heterostructure in which electronic characteristics can be tuned as per the requirement of a particular application. Such laterally integrated hybrid nanostructures are investigated using density functional theory to explore their growth, electronic properties, and chemical topology. The evolution of energetics, geometry, density of states, number of charges, and electric dipole moment with carbon adatoms is delineated. In the triangular C–BN heterostructures, C atoms prefer to grow from the vertices as compared to the edges of the heterostructures due to their strong anchoring. Despite a small lattice mismatch between h-BN and graphene, the structural stability of the heterostructure depends on the number of C adatoms (i.e. stage of growth), and growth from the B-terminated BN flake appears to be preferred. The Cu substrate reduces the stability of the C–BN heterostructure. The heterostructures are metallic, suggesting that charge transfer effects from the Cu substrate play a dominant role in governing the electronic properties of the heterostructures. Electron localization and quantum theory of atoms in molecules illustrate the partial ionic–covalent character of the B–N bonds, which are less covalent than C–B/C–N bonds in the heterostructures. All in all, this study on the evolution of the characteristics of the C–BN heterostructures with growth is a vital step towards developing hybrid nanoelectronic circuitry with precisely controlled properties.
- This article is part of the themed collection: Interdisciplinary Symposium on Materials Chemistry 2014
 Please wait while we load your content...
Please wait while we load your content...

