
Molecular dynamics simulations on lithium diffusion in LiFePO4: the effect of anti-site defects†
Abstract
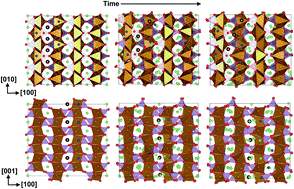
As a promising cathode material for rechargeable lithium ion batteries, lithium iron phosphate (LiFePO4) is prevented from further implementations due to its low ionic and electronic conductivity. Anti-site defects were reported earlier to be responsible for the slow lithium ion diffusion. However, the underlying mechanism is controversial. In this work we perform molecular dynamics simulations to investigate the real time diffusion dynamics of lithium ions in LiFePO4 containing Li–Fe anti-site defects. Our results indicate that the anti-site defects indeed slow down Li-ion diffusion in [010] channels. However as the concentration of anti-site defects increases, Li-ion diffusion becomes more isotropic. A local structural transition from the solid phase to a liquid-like solid solution in LiFePO4 with anti-site defects is observed around the defect concentration of 8%, which coincides with the dynamical transition from the anisotropic 1D diffusion of Li ions to the isotropic 2D or 3D diffusion. At high temperatures, the anti-site defects could even enhance Li diffusion. The defect distribution also plays an important role in modulating Li diffusion. The recombination of the anti-site defect and the inter-channel crossing are observed for the edge-shared defect model. Furthermore the aggregation of defects could induce quite different local environments, resulting in heterogeneous diffusion, which may reconcile the theoretical predictions on the Li diffusion coefficient with the experimental measurements.
 Please wait while we load your content...
Please wait while we load your content...

