
Benchmarking density functional theory predictions of framework structures and properties in a chemically diverse test set of metal–organic frameworks†
Abstract
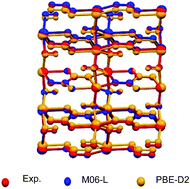
A test set of chemically and topologically diverse Metal–Organic Frameworks (MOFs) with high accuracy experimentally derived crystallographic structure data was compiled. The test set was used to benchmark the performance of Density Functional Theory (DFT) functionals (M06L, PBE, PW91, PBE-D2, PBE-D3, and vdW-DF2) for predicting lattice parameters, unit cell volume, bonded parameters and pore descriptors. On average PBE-D2, PBE-D3, and vdW-DF2 predict more accurate structures, but all functionals predicted pore diameters within 0.5 Å of the experimental diameter for every MOF in the test set. The test set was also used to assess the variance in performance of DFT functionals for elastic properties and atomic partial charges. The DFT predicted elastic properties such as minimum shear modulus and Young's modulus can differ by an average of 3 and 9 GPa for rigid MOFs such as those in the test set. The partial charges calculated by vdW-DF2 deviate the most from other functionals while there is no significant difference between the partial charges calculated by M06L, PBE, PW91, PBE-D2 and PBE-D3 for the MOFs in the test set. We find that while there are differences in the magnitude of the properties predicted by the various functionals, these discrepancies are small compared to the accuracy necessary for most practical applications.
 Please wait while we load your content...
Please wait while we load your content...

