
Initial decomposition reaction of di-tetrazine-tetroxide (DTTO) from quantum molecular dynamics: implications for a promising energetic material†
Abstract
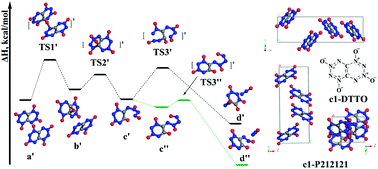
Di-tetrazine-tetroxide (DTTO) was predicted in 2001 to have a density (up to 2.3 g cm−3) and heat of detonation (up to 421.0 kcal mol−1) better than other explosives, making it the “holy grail” of energetic materials (EMs), but all attempts at synthesis have failed. We report Density Functional Theory (DFT) molecular dynamics simulations (DFT-MD) on DTTO crystal for the two most stable monomers. We predict that the most stable isomer (c1) has a density of ρ = 1.96 g cm−3 with an estimated detonation velocity (Dv) of 9.70 km s−1 and a detonation pressure (Dp) of 43.0 GPa, making it comparable to RDX (ρ = 1.82 g cm−3, Dv = 8.75 km s−1, Dp = 35.0 GPa), HMX (ρ = 1.91 g cm−3, Dv = 9.10 km s−1, Dp = 39.3 GPa) and CL-20 (ρ = 2.04 g cm−3, Dv = 9.38 km s−1, Dp = 44.1 GPa). The DFT-MD studies find that the initial reaction at lower pressure is unimolecular decomposition to form two N2O molecules (barrier 45.9 kcal mol−1), while at higher pressure it is intermolecular oxygen-transfer with a barrier of 40.1 kcal mol−1. For the c2 isomer (less stable by 1.2 kcal mol−1) the initial reaction involves two DTTO molecules reacting to form a dimer which then releases N2 as a direct product (barrier 48.1 kcal mol−1), a unique initial reaction among EMs. These results suggest that DTTO may have a higher thermal stability (barrier >7.0 kcal mol−1 higher) than RDX, HMX, and CL-20.
 Please wait while we load your content...
Please wait while we load your content...

