
Thermodynamics of the self-assembly of non-ionic chromonic molecules using atomistic simulations. The case of TP6EO2M in aqueous solution†
Abstract
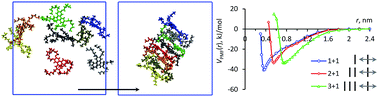
Atomistic molecular dynamic simulations have been performed for the non-ionic chromonic liquid crystal 2,3,6,7,10,11-hexa-(1,4,7-trioxa-octyl)-triphenylene (TP6EO2M) in aqueous solution. TP6EO2M molecules consist of a central poly-aromatic core (a triphenylene ring) functionalized by six hydrophilic ethyleneoxy (EO) chains, and have a strong tendency to aggregate face-to-face into stacks even in very dilute solution. We have studied self-assembly of the molecules in the low concentration range corresponding to an isotropic solution of aggregates, using two force fields GAFF and OPLS. Our results reveal that the GAFF force field, even though it was successfully used previously for modelling of ionic chromonics, overestimates the attraction of TP6EO2M molecules in water. This results in an aggregation free energy which is too high, a reduced hydration of EO chains and, therefore, molecular self-assembly into compact disordered clusters instead of stacks. In contrast, use of the OPLS force field, leads to self-assembly into ordered stacks in agreement with earlier experimental studies of triphenylene-based chromonics. The free energy of association follows a “quasi-isodesmic” pattern, where the binding free energy of two molecules to form a dimer is of the order of 2.5 RT larger than the corresponding energy of addition of a molecule into a stack. The obtained value for the binding free energy, ΔG0agg = −12 RT, is found to be in line with the published values for typical ionic chromonics (−7 to −12 RT), and agrees reasonably well with the experimental results for this system. The calculated interlayer distance between the molecules in a stack is 0.37 nm, which is at the top of the range found for typical chromonics (0.33–0.37 nm). We suggest that the relatively large layer spacing can be attributed to the repulsion between EO side chains.
 Please wait while we load your content...
Please wait while we load your content...

