
Influence of enolate/epoxy configuration, doping and vacancy on the catalytic activity of graphene†
Abstract
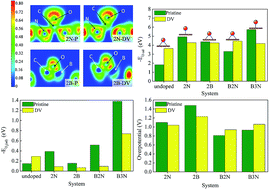
Using density functional theory based electronic structure analysis we substantiate that the bonding type of atomic oxygen (epoxide or enolate) and adsorption strength of molecular oxygen play vital roles in determining the overpotential of oxygen reduction reactions (ORR) of n and p doped graphene-based electrocatalysts. The presence of localized pz states influences the electron accepting and donating characteristics of the carbon atoms of the DV (555-777) defective graphene. We probe the origin of dopant-induced enolate and epoxide formation in both pristine and DV graphene based on the occupation of pz orbital of active site after doping. In spite of the slightly higher tendency of DV to adsorb molecular oxygen than pristine graphene, the enhancement in binding strengths of O2 with the introduction of dopants is higher in the pristine case when compared to the DV. The donation and back-donation interaction of dopant with nearby carbon atoms is inspected in detail. We examine the effects of boron and nitrogen co-doping and find that a doping configuration of a boron atom bonded to two nitrogen atoms (B2N) possesses moderate binding with atomic and molecular oxygen, suggesting it to be a better catalyst for oxygen reduction reaction with lowest overpotential among the systems considered. Moreover the possibility of CO poisoning has been tested for all the surfaces. The approach considered in this work is general and can be extended to understand the catalytic activity of other carbon/graphene based systems.
 Please wait while we load your content...
Please wait while we load your content...

