
The applicability of the dimeric heterosynthon concept to molecules with equivalent binding sites. A DFT study of crystalline urea–H2O2†
Abstract
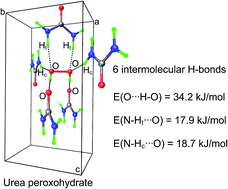
The limited applicability of the dimeric heterosynthon concept to a two-component urea–H2O2 crystal is reported. It is due to the absence of the relatively short O–H⋯O bonds, i.e. primary interactions, in the urea–H2O2 1 : 1 complex. The target O–H⋯O bonds do exists in trimeric heterosynthons, i.e. the urea–2(H2O2) and 2(urea)–H2O2 complexes. The mutual orientation of the H2O2 molecules in the gas-phase complexes differs from the one in the crystalline structure due to the existence of additional N–H⋯O bonds which are absent in the crystal. Implicitly accounting for the polar environment does not change the molecule conformations in the considered complexes. It is found that the DFT computations with/without accounting for polar solvent are not sufficient for the deduction of such a heterosynthon. The results of the database analysis should be used for unambiguous identification of the molecules' conformations in the target trimeric heterosynthon. An approach for deducing the trimeric heterosynthon structure for molecules with equivalent binding sites is developed. It includes three steps. (i) Identification of structural motifs formed by the considered molecules in the two-component crystals using database analysis. (ii) Establishing a hierarchy of the intermolecular interactions in the crystals by solid-state DFT followed by Bader analysis of the periodic electronic density. (iii) Evaluation of the structure and relative stability of the trimeric heterosynthons by DFT methods with/without accounting for environmental effects.
 Please wait while we load your content...
Please wait while we load your content...

